

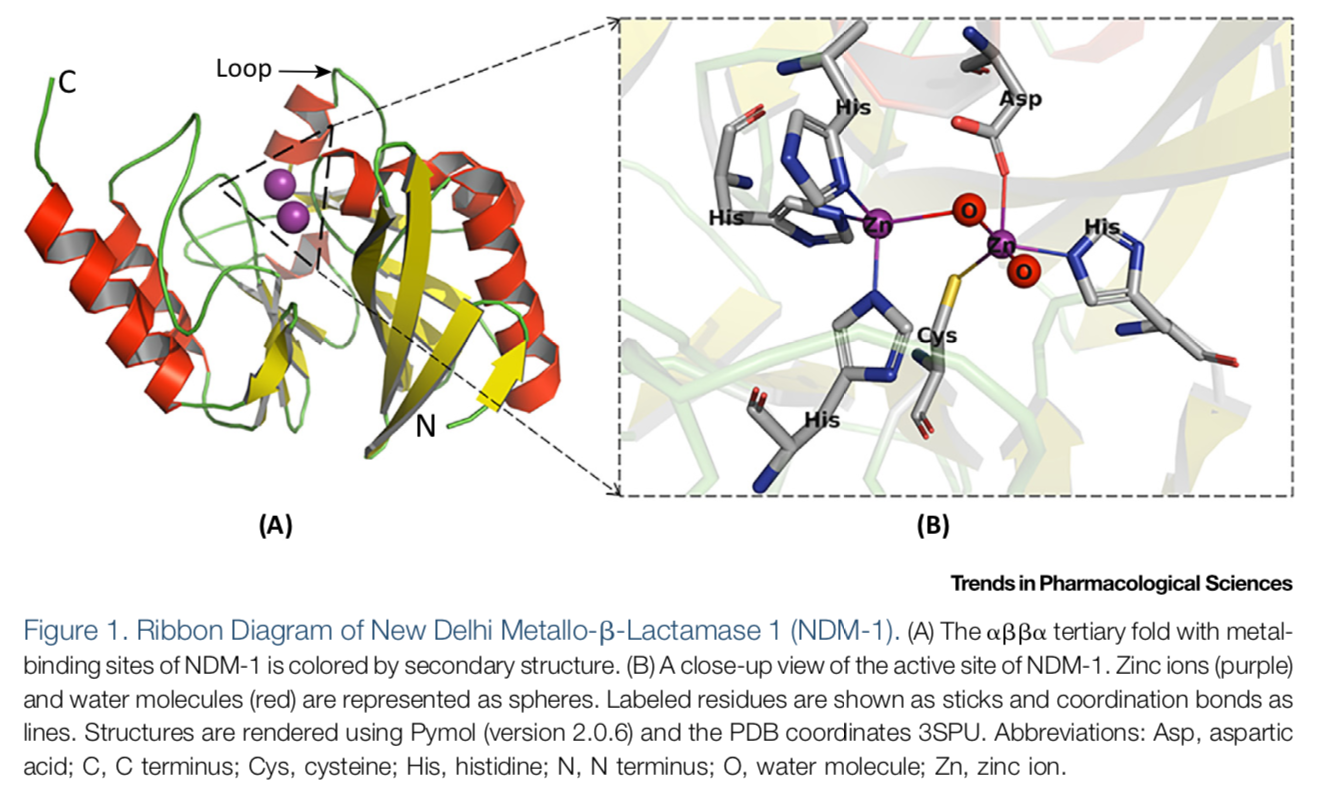
Bacterial infections are most commonly treated by the use of β-lactam antibiotics. β-Lactam antibiotics are capable of inhibiting transpeptidases , penicillin-binding proteins (PBPs), involved in cell wall biosynthesis1. Approximately two million people in the United States each year acquire bacterial infections that are resistant to one or more antibiotics and about 23,000 people die due to this resistance1. Resistance to β-lactam antibiotics has become increasingly prevalent since the introduction of antibiotics2. Resistance to β-lactam antibiotics has become increasingly prevalent since the introduction of antibiotics. A common mechanism for β-lactam resistance is the production of β-lactamases, which hydrolyze the β-lactam ring, thus rendering the drugs inactive. Today there are more than 2,800 β-lactamases, which are classified into four groups according to molecular properties and/or amino acid sequences: A, B, C and D 3 . The group A, C, and D β- lactamases use an active site serine for catalysis, while the group B β-lactamases use one or two Zn(II) ions and are called metallo-β-lactamases (MBLs). The most clinically-relevant MBLs are of the B1 subclass and include the following: IMiPenemase (IMP-); New Delhi Metallo-β- lactamase (NDM-); and Verona Integrin-encoded Metallo-β-lactamase (VIM-).
 (See permission in related links)
(See permission in related links)
The emergence of new clinical variants of MBLs continues to be an ongoing battle; currently there are 80 clinical variants of IMP; 66 variants of VIM; and 28 variants of NDM 4 . Selective pressures continue to drive the evolution of MBLs with the clinical variants showing improvements in stability, selectivity, and/or kinetics properties. For example, some MBLs have evolved to require only one Zn(II) for full catalytic activity and maintain the ability to hydrolyze carbapenems. Recently, it has also been found that clinical variants of NDM and VIM have evolved to have improved activity at low zinc concentrations 5 6 . In addition, MBL genes are typically encoded on mobile elements, with some MBL-encoding plasmids carrying resistance genes for other classes of antibiotics 7 . This trait increases the difficulty of effective treatment for bacterial infections.
References
1. Neu, H. C., beta-Lactam antibiotics: structural relationships affecting in vitro activity and pharmacologic properties. Rev Infect Dis 1986, 8 Suppl 3, S237-59.
2. CDC, Antibiotic Resistance Threats in the Unitied States. 2019.
3. Tooke, C. L.; Hinchliffe, P.; Bragginton, E. C.; Colenso, C. K.; Hirvonen, V. H. A.; Takebayashi, Y.; Spencer, J., beta-Lactamases and beta-Lactamase Inhibitors in the 21st Century. J Mol Biol 2019, 431 (18), 3472-3500.
4. Naas, T.; Oueslati, S.; Bonnin, R. A.; Dabos, M. L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B. I., Beta-lactamase database (BLDB) – structure and function. Journal of Enzyme Inhibition and Medicinal Chemistry 2017, 32 (1), 917-919.
5. Cheng, Z.; Thomas, P. W.; Ju, L.; Bergstrom, A.; Mason, K.; Clayton, D.; Miller, C.; Bethel, C. R.; VanPelt, J.; Tierney, D. L.; Page, R. C.; Bonomo, R. A.; Fast, W.; Crowder, M. W., Evolution of New Delhi metallo-beta-lactamase (NDM) in the clinic: Effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J Biol Chem 2018, 293 (32), 12606-12618.
6. Cheng, Z.; Shurina, B. A.; Bethel, C. R.; Thomas, P. W.; Marshall, S. H.; Thomas, C. A.; Yang, K.; Kimble, R. L.; Montgomery, J. S.; Orischak, M. G.; Miller, C. M.; Tennenbaum, J. L.; Nix, J. C.; Tierney, D. L.; Fast, W.; Bonomo, R. A.; Page, R. C.; Crowder, M. W., A Single Salt Bridge in VIM-20 Increases Protein Stability and Antibiotic Resistance under Low-Zinc Conditions. mBio 2019, 10 (6).
7. Bush, K., Past and Present Perspectives on beta-Lactamases. Antimicrob Agents Chemother 2018, 62 (10).
The main factor driving the development of MBL inhibitors is the lack of effective treatments to overcome MBL-mediated resistance in the clinic. In the past 5 years, there has been significant effort to identify novel inhibitors of the MBL. The number of MBL inhibitors and publications describing MBL inhibitors have increased dramatically over the last decade, with over 900 inhibitors reported in literature. Many studies group inhibitors by their structural characteristics 1 2 3 , but an alternative categorization may be more useful and that is grouping by mechanism of inhibition 4 5 . After researching MBL inhibitors reported in the literature, it was determined that most structural categories can be grouped into one of four different mechanisms: inhibition through metal ion binding; inhibition through covalent bond formation; allosteric inhibitors; and inhibitors with uncharacterized mechanisms 4 .
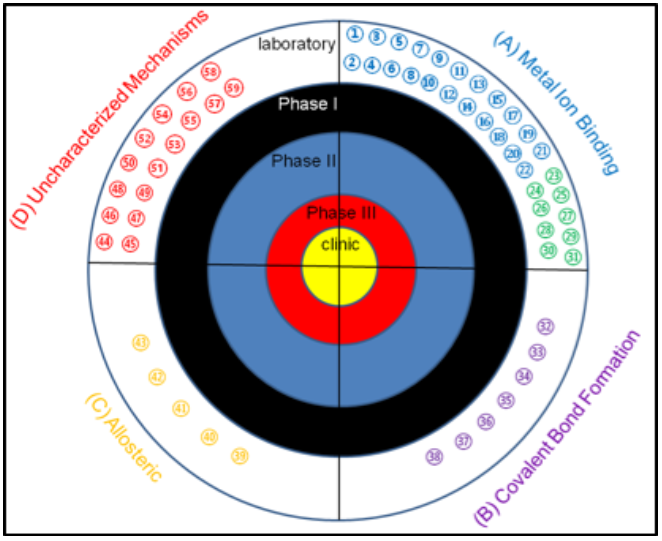 Target for MBL inhibitors. Previously-reported MBL inhibitors were classified into 4 groups: MBL inhibitors that
target the metal ions, MBL inhibitors that form covalent adducts, MBL inhibitors that bind to sites away from the
active site, and MBL inhibitors with unknown mechanism of inhibition. To date, there are no MBL inhibitors in the
clinic or in any clinical trials.REF (1) 1,2,4-triazole-3-thiones, (2) captopril and analogs, (3) cyclic boronates,
(4) bisthiazolidines, (5) mercaptoacetate, (6) mercaptocarboxylate (7) mercaptophosphonates, (8) aminophtalic
acid derivatives, (9) disubstituted succinic acids, (10) biphenyl tetrazoles, (11) tiopronin, (12) pyridine-2,
4-dicarboxylic acids, (13) thiomandelic acids, other thiol containing derivatives, (14) rhodamine hydrolysis
products, (15) triazolythioacetamide analog, (16) sulfonamides, (17) tricyclic natural products, (18) benzophenone,
(19) 3-oxoisoindoline-4-carboxylates, (20) dipicolinic acid derivatives, (21) benzyl thiol, and (22) cyclobutanone
penem analog. (23) EDTA, (24) 1,10-phenanthroline, (25) picolinic acids, (26) TPEN, (27) NODAGA, (28) NOTA, (29) DOTA,
(30) 1,4,7-triazonane-1,4,7-tris (carboxylodithioate) analogs, (31) AMA, (32) mercaptophenylacetic acid, (33)
moxalactam, (34) cephamycins, (35) 3-(3-mercaptopropionylsulfanyl)- propionic acid pentafluorophenyl ester, (36)
cefaclor, (37) ebselen, (38) formylchromone.(39) arginine peptides, (40) camelid nanobody, (41) graphene oxide
and nanotubes, (42) DNA nanoribbon, (43) single stranded DNA (44) azolylthioacetamides, (45) several different
classes of thionylpeptides, (46) hydroxamates and “reverse” hydroxamates, (47) maleic acids, (48) mitoxantrone,
(48) mitoxantrone, (49) triazoles, (50) mercaptotriazoles, (51) substituted thiosemicarbazides, (52) arylsulfonyl
hydrozones, (53) pyrrole derivatives, (54) thioesters, (55) thiols, (56) trifluoro alcohols and ketones, (57)
sulfonamides, (58) polyketides, and (59) cystatins
Target for MBL inhibitors. Previously-reported MBL inhibitors were classified into 4 groups: MBL inhibitors that
target the metal ions, MBL inhibitors that form covalent adducts, MBL inhibitors that bind to sites away from the
active site, and MBL inhibitors with unknown mechanism of inhibition. To date, there are no MBL inhibitors in the
clinic or in any clinical trials.REF (1) 1,2,4-triazole-3-thiones, (2) captopril and analogs, (3) cyclic boronates,
(4) bisthiazolidines, (5) mercaptoacetate, (6) mercaptocarboxylate (7) mercaptophosphonates, (8) aminophtalic
acid derivatives, (9) disubstituted succinic acids, (10) biphenyl tetrazoles, (11) tiopronin, (12) pyridine-2,
4-dicarboxylic acids, (13) thiomandelic acids, other thiol containing derivatives, (14) rhodamine hydrolysis
products, (15) triazolythioacetamide analog, (16) sulfonamides, (17) tricyclic natural products, (18) benzophenone,
(19) 3-oxoisoindoline-4-carboxylates, (20) dipicolinic acid derivatives, (21) benzyl thiol, and (22) cyclobutanone
penem analog. (23) EDTA, (24) 1,10-phenanthroline, (25) picolinic acids, (26) TPEN, (27) NODAGA, (28) NOTA, (29) DOTA,
(30) 1,4,7-triazonane-1,4,7-tris (carboxylodithioate) analogs, (31) AMA, (32) mercaptophenylacetic acid, (33)
moxalactam, (34) cephamycins, (35) 3-(3-mercaptopropionylsulfanyl)- propionic acid pentafluorophenyl ester, (36)
cefaclor, (37) ebselen, (38) formylchromone.(39) arginine peptides, (40) camelid nanobody, (41) graphene oxide
and nanotubes, (42) DNA nanoribbon, (43) single stranded DNA (44) azolylthioacetamides, (45) several different
classes of thionylpeptides, (46) hydroxamates and “reverse” hydroxamates, (47) maleic acids, (48) mitoxantrone,
(48) mitoxantrone, (49) triazoles, (50) mercaptotriazoles, (51) substituted thiosemicarbazides, (52) arylsulfonyl
hydrozones, (53) pyrrole derivatives, (54) thioesters, (55) thiols, (56) trifluoro alcohols and ketones, (57)
sulfonamides, (58) polyketides, and (59) cystatins
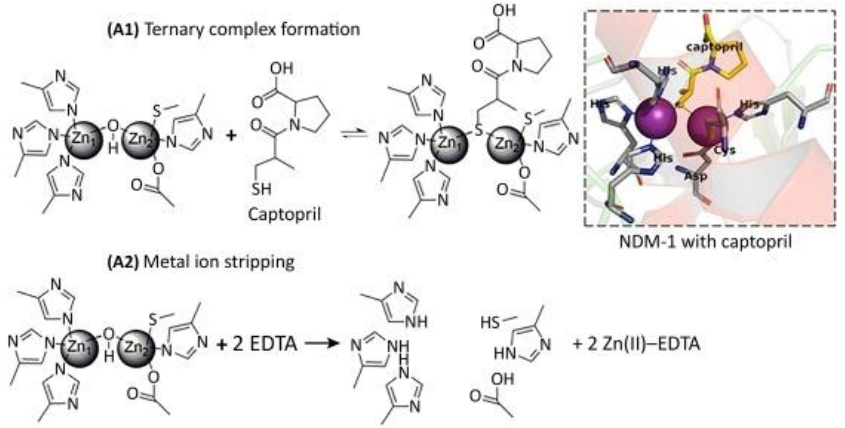
Inhibitors that act through metal ion binding may be further sub-classified by mechanism of action. Metal ion stripping suggests that the inhibitor performs one of two actions: (i) the inhibitor removes the metal ions from the enzyme’s active site; or (ii) the inhibitor isolates and binds to metal ions that are not bound to the active site. If the metal-ion-binding inhibitor does not strip the enzyme of its metal ions, then it forms a ternary complex with the enzyme wherein the inhibitor binds to the metal ions and neighboring amino acids in order to prevent the enzyme from binding to antibiotics.
(See permission in related links)
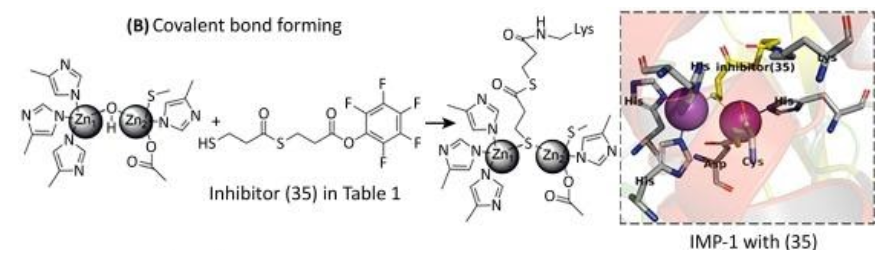
With regards to inhibition through covalent bond formation, the inhibitor covalently binds to the enzyme resulting in irreversible inhibition. These inhibitors can overcome high substrate concentrations with reasonable efficiency. Covalent bond-forming inhibitors prove to be inconvenient due to the availability of a common amino acid residue for modification.
(See permission in related links)
Allosteric inhibitors act by binding to an exosite on the enzyme in order to decrease the enzyme’s binding affinity for the substrate (β-lactams) or decrease enzyme activity. Inhibitors of this variety are generally macromolecules that present challenges in characterization and production. Because MBLs have such a great variance in sequences outside of the active site, broad-spectrum inhibitors would be extremely difficult to obtain.
(See permission in related links)
Inhibitors with uncharacterized mechanisms are normally classified solely upon their inhibitory concentration at half-maximum (IC50) values. This sole classification is assumed to be inadequate for proceeding with inhibitor development.
References
1. Fast, W.; Sutton, L. D., Metallo-beta-lactamase: inhibitors and reporter substrates. Biochim Biophys Acta 2013, 1834 (8), 1648-59.
2. McGeary, R. P.; Tan, D. T.; Schenk, G., Progress toward inhibitors of metallo-beta-lactamases. Future Med Chem 2017, 9 (7), 673-691.
3. Rotondo, C. M.; Wright, G. D., Inhibitors of metallo-beta-lactamases. Curr Opin Microbiol 2017, 39, 96-105.
4. Ju, L. C.; Cheng, Z.; Fast, W.; Bonomo, R. A.; Crowder, M. W., The Continuing Challenge of Metallo-beta-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol Sci 2018, 39 (7), 635-647.
5. Shi, C.; Chen, J.; Kang, X.; Shen, X.; Lao, X.; Zheng, H., Approaches for the discovery of metallo-beta-lactamase inhibitors: A review. Chem Biol Drug Des 2019, 94 (2), 1427-1440.